-
Multi-scale modelling of solvatochromic shifts from frozen-density embedding theory with non-uniform continuum model of the solvent: the coumarin 153 case
X. Zhou, J.W. Kaminski and T.A. Wesolowski
Physical Chemistry Chemical Physics, 13 (22) (2011), p10565-10576


DOI:10.1039/C0CP02874F | unige:15943 | Abstract | Article HTML | Article PDF
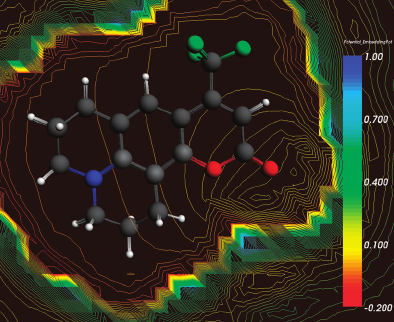
For nine solvents of various polarity (from cyclohexane to water), the solvatochromic shifts of the lowest absorption band of coumarin 153 are evaluated using a computational method based on frozen-density embedding theory [Wesolowski and Warshel, J. Chem Phys., 1993, 97, 9050, and subsequent articles]. In the calculations, the average electron density of the solvent ãÏB(râ)ã is used as the frozen density. ãÏB(râ)ã is evaluated using the statistical-mechanical approach introduced in Kaminski et al., J. Phys. Chem. A, 2010, 114, 6082. The small deviations between experimental and calculated solvatochromic shifts (the average deviation equals to about 0.02 eV), confirm the adequacy of the key approximations applied: (a) in the evaluation of the average effect of the solvent on the excitation energy, using the average density of the solvent instead of averaging the shifts over statistical ensemble and (b) using the approximant for the bi-functional of the non-electrostatic component of the orbital-free embedding potential, are adequate for chromophores which interact with the environment by non-covalent bonds. The qualitative analyses of the origin of the solvatochromic shifts are made using the graphical representation of the orbital-free embedding potential.